
WhichTF is dominant in your open chromatin data?
Published in PLOS Comput Biol, 2022
We developed an ontology-guided approach to ranking tissue-/cell-type-specific transcription factors (TFs) from chromatin accessibility data.
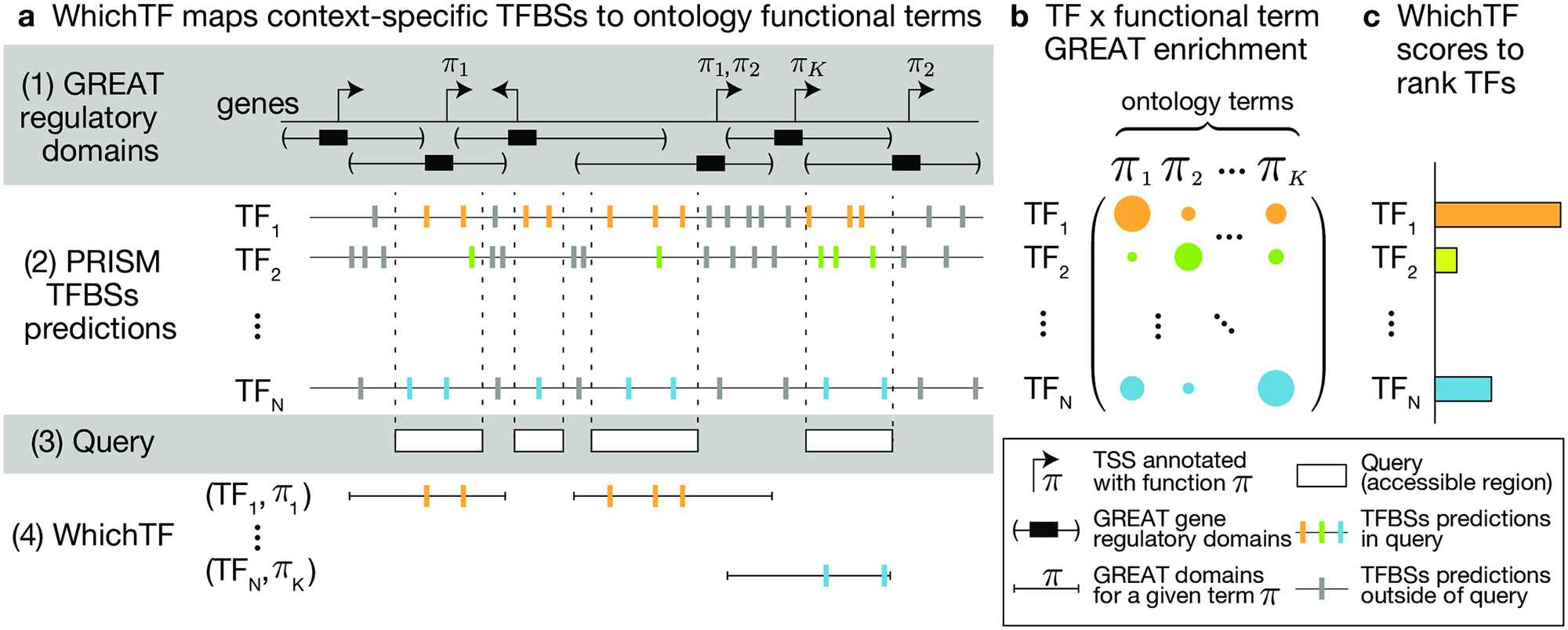
We have presentation slides on this work here.
Highlights
Transcription factors (TFs) regulate context-specific gene expression. Genome-wide chromatin accessibility profiles coupled with computational analysis have been some of the most promising approaches to rank TFs enriched in a given sample. Most existing methods rely on statistical enrichment, either on sequence motifs or occurrences of the TF binding sites. We empirically find that these abundance-based approaches tend to be less cell-type-specific. For example, CTCF is frequently ranked among the top hits.
To address this, we propose an alternative, WhichTF, that aggregates the results of ontology-based stratified enrichment tests to rank TFs. Our model considers ontology-based annotations of genomic regions on top of high-confidence predictions of conserved TF binding sites. Specifically, we decompose the user-provided accessible profiles into a series of genomic tracks annotated for ontology terms. We consider the most enriched terms in the accessible peaks as a proxy to capture cellular contexts. By aggregating across terms, we score and rank TFs.
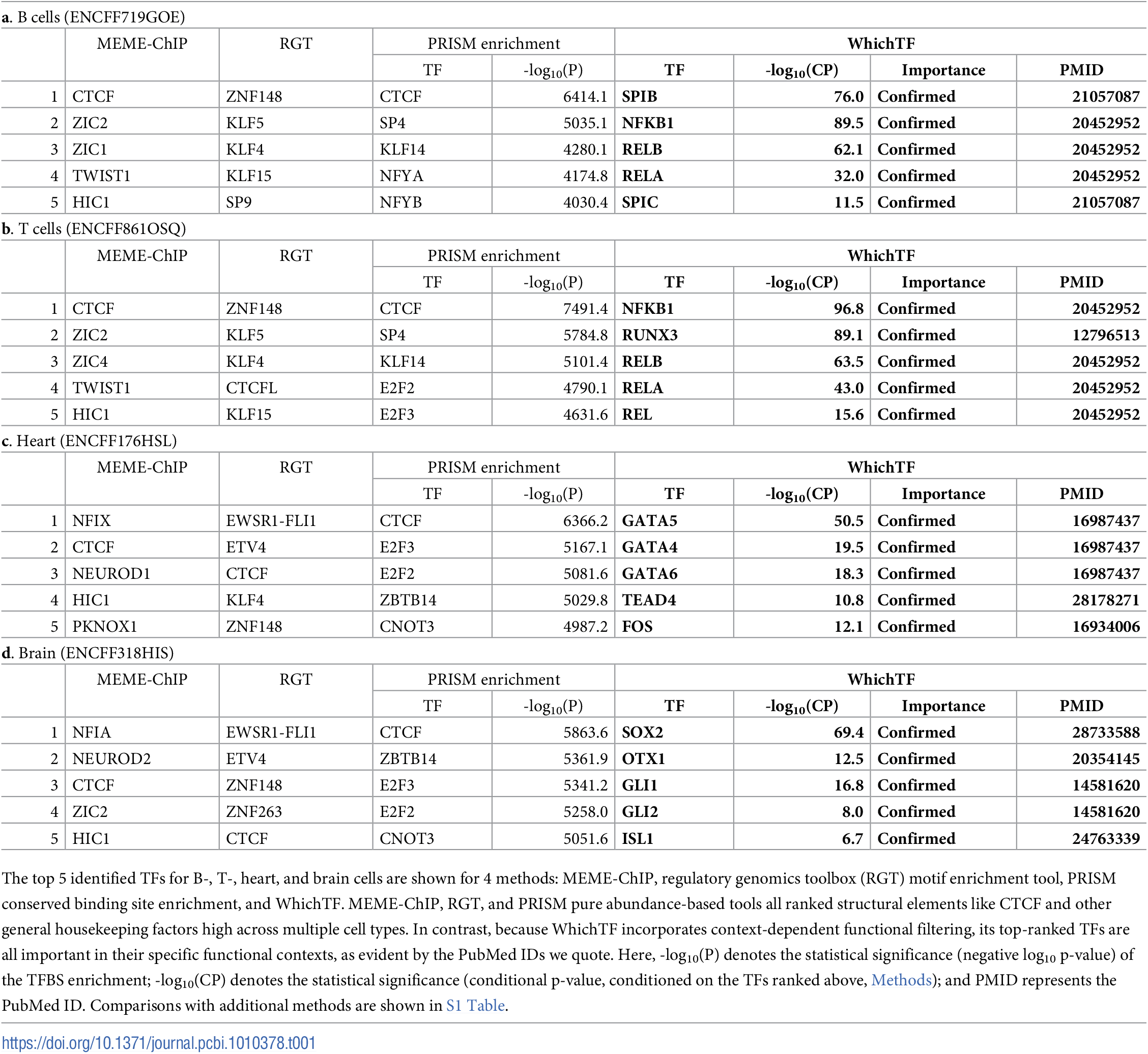
When applied to well-characterized datasets, we find that the cell-type-specific roles of the top-ranked TFs are supported by the literature.
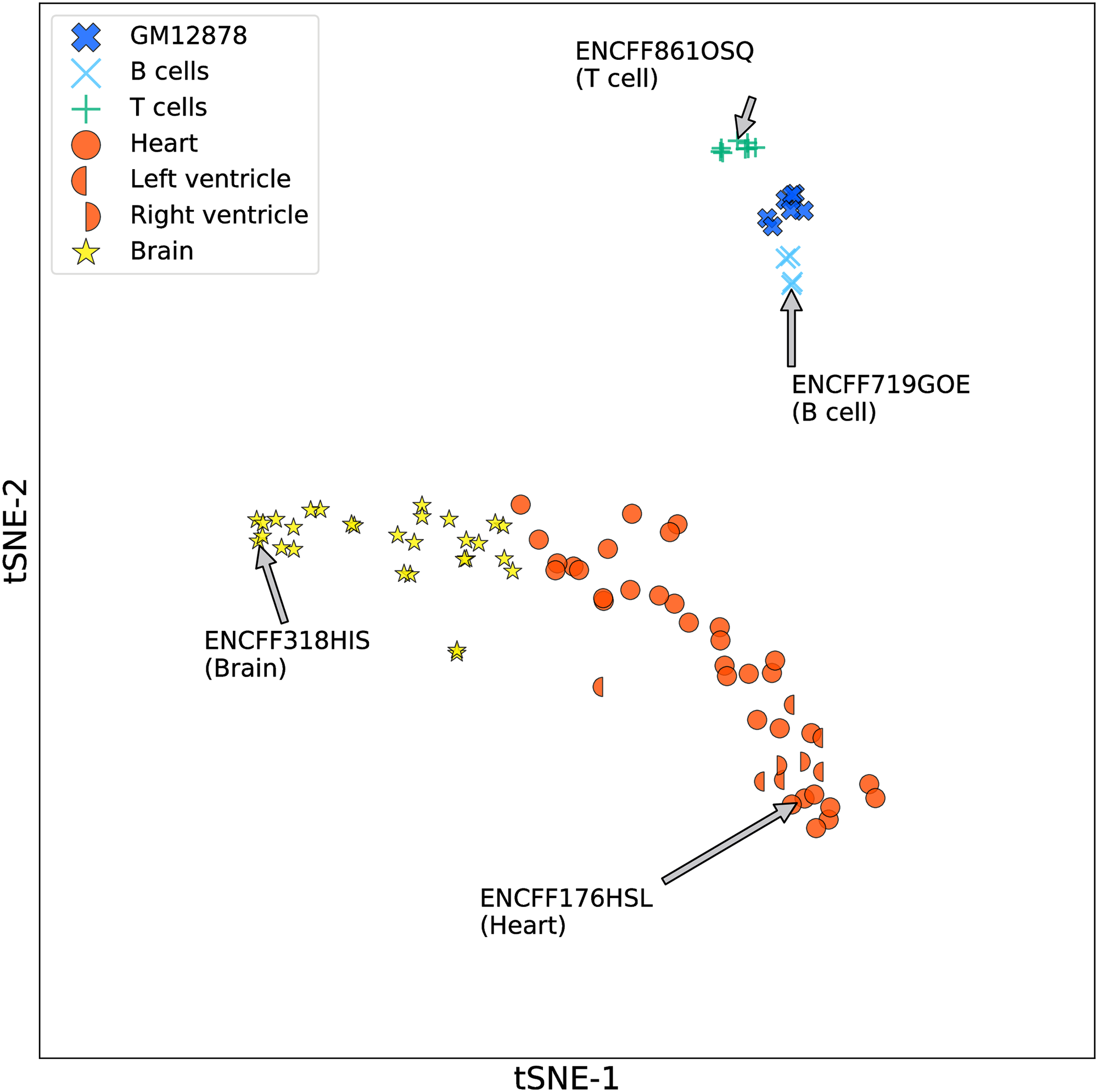
Applying across ~90 human samples followed by the t-SNE projection of the WhichTF scores TFs indicates WhichTF captures biological similarities and dissimilarities of TF-mediated transcriptional programs.
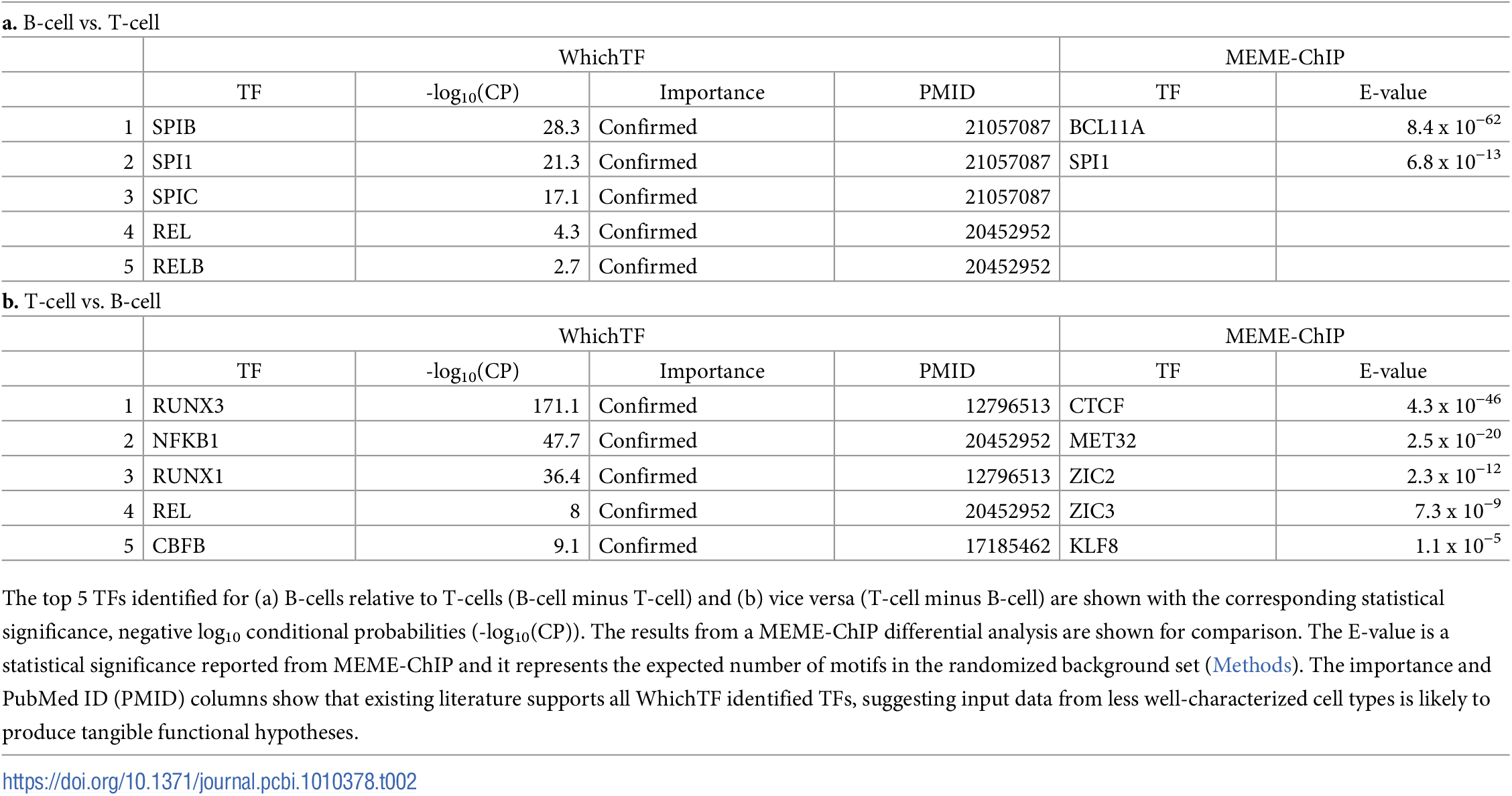
To investigate closely related cells, we present differential TF ranking with WhichTF, where we apply WhichTF to differentially accessible regions. The top-ranked TFs are well-supported by the literature.
Beyond well-characterized cell types, we present applications in mesoderm development,
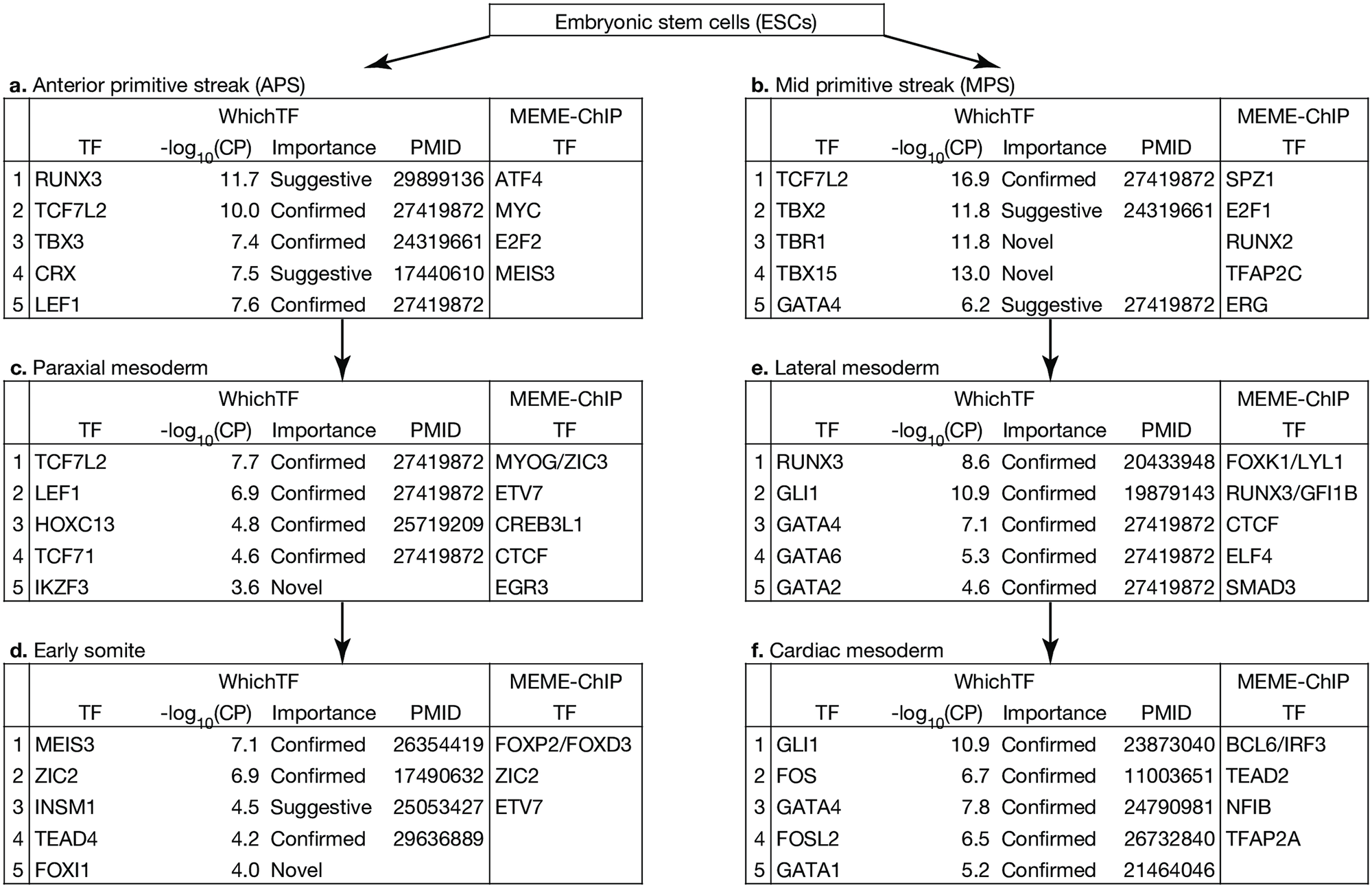
… and patient-derived samples for systemic lupus erythematosus.
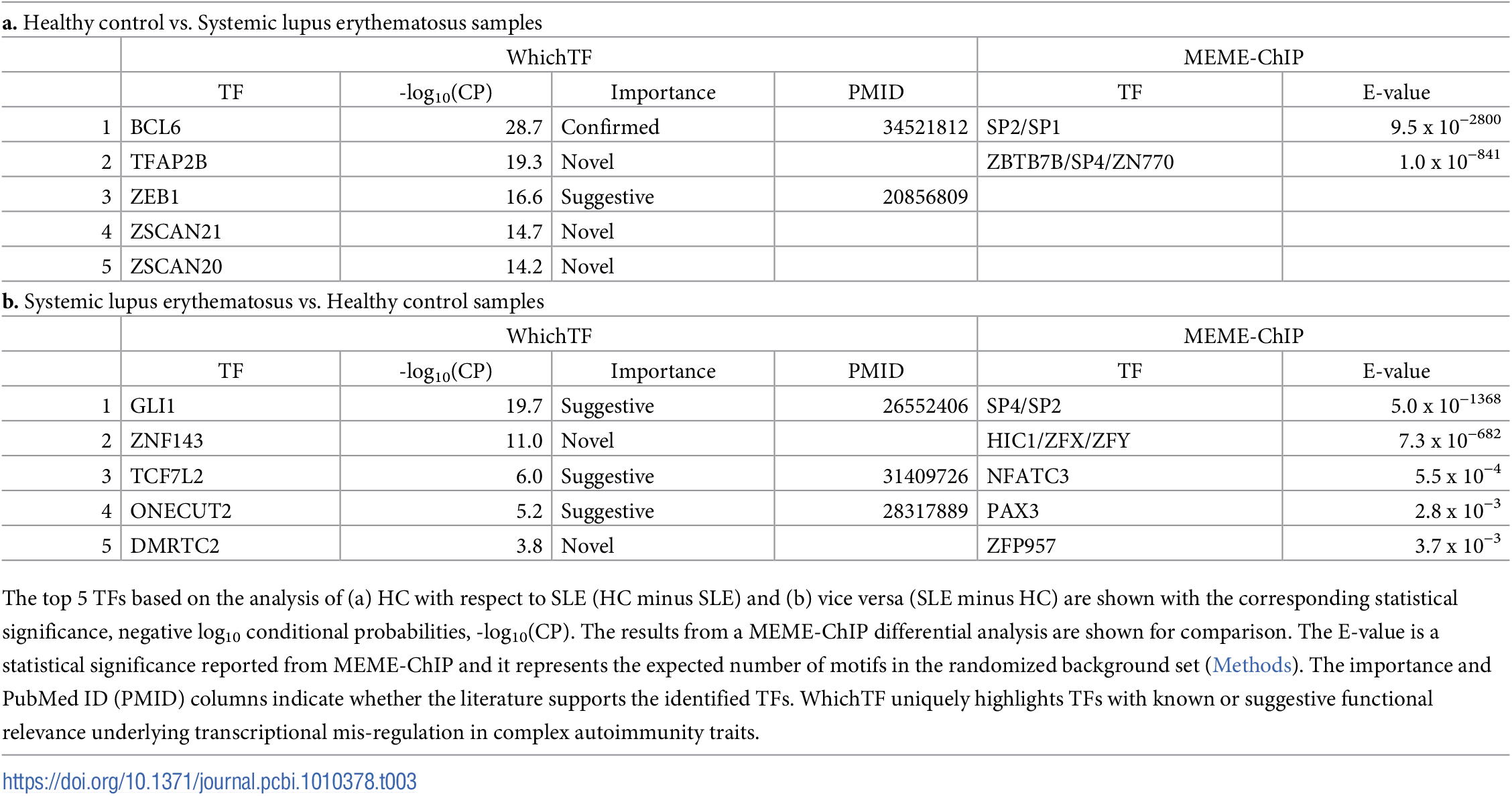
Overall, we show that ontology-guided stratified enrichment is a powerful approach for ranking cell-type-specific TFs. We highlight examples in human and mouse cells, developmental trajectories, and disease samples.
This work was started and was jointly led by Ethan S. Dyer and myself while I was in Prof. Gill Bejerano’s lab at Stanford, with tremendous support and help from many lab members. I want to thank Gill, Ethan, and my colleagues. Thank you, all!
Resources
WhichTF software and reference data are available on BitBucket and figshare.
Also, as a part of this project, we performed a v4 update of the Genomic Regions Enrichment of Annotations Tool (GREAT).
日本語での解説 (Highlights in Japanese)
転写因子とよばれるタンパク質は、DNAに対して配列特異的に結合し近傍の遺伝子の発現を調節する。がん細胞を除く全身の細胞で同一のゲノム情報が共有されているものの、組織・細胞種特異的に遺伝子発現が行われるのは、転写因子による調節によるところが大きいとされている。もし、特定の組織・細胞においてどの転写因子が重要であるかが特定できていれば、その転写因子のDNAとの結合状態をゲノム全体において実験的に調べることができるが、1500種類もある転写因子のなかで、どれが重要であるかを特定することは困難である。
本論文では、WhichTFというバイオ・インフォマティクス・ツールを開発し、細胞種特異的な機能に関わり重要である転写因子を特定することに有用であることを示した。提案手法は、重要であると推定される転写因子の順位付きリストを求めるために、次に挙げられる複数の情報を組み合わている:ユーザーにより提供される、細胞種特異的なオープンクロマチン領域の情報、哺乳類におけるゲノムの進化的保存度の情報、過去の生物医学研究により得られたオントロジーに基づくゲノム注釈の情報、そして、転写因子により認識されるゲノム配列特徴の情報。ベンチマーク実験を行ったところ、これらの情報源の一部しか用いない既存の手法は、細胞種に関わらずに重要な役割を果たすCTCFなどクロマチン構造因子をトップヒットとすることがわかり、提案手法による統合解析の優位性が示された。
次に、私たちはWhichTFが多くの細胞に対して適用可能なことを示した。ヒトとマウスの培養細胞、ヒトの中胚葉発生、さらには疾患細胞(全身性エリテマトーデスの患者由来サンプル)においても、WhichTFにより重要であると推定される転写因子の多くに、機能を示した既存の論文があること、また、WhichTFを用いることでこれまでに示唆されていない転写因子を同定できることを示した。
提案手法を活用することで、発生・疾患などの多様な細胞において重要な転写因子を同定し、発生や疾患にともなう細胞内での遺伝子発現制御のメカニズムに関する理解が進展することが期待される。
Reference: Y. Tanigawa*, E. S. Dyer*, G. Bejerano, WhichTF is dominant in your open chromatin data? PLOS Comput Biol 18(8): e1010378. (2022). https://doi.org/10.1371/journal.pcbi.1010378